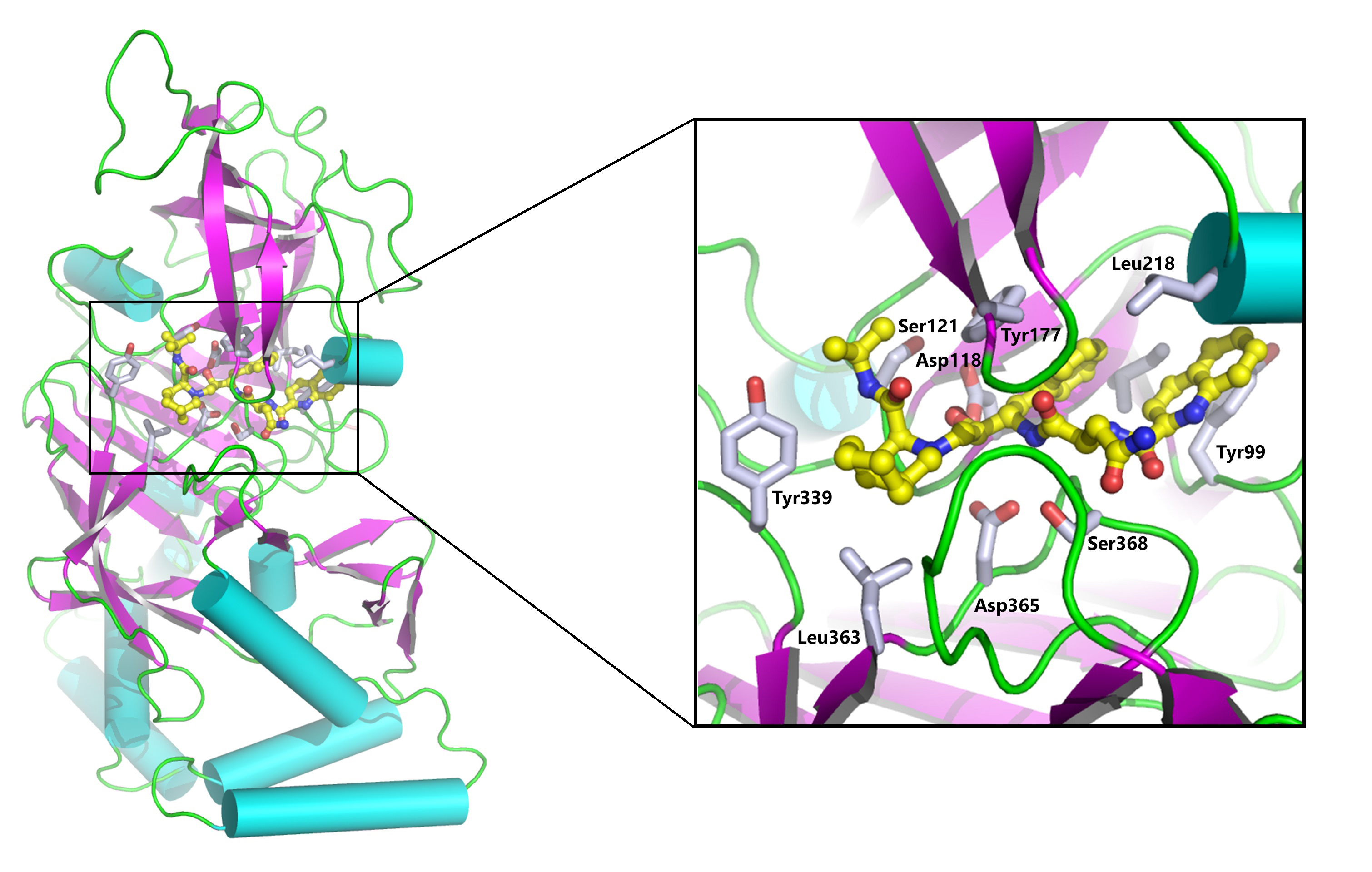
Structural studies on substrate binding protein
Periplasmic substrate binding proteins (SBPs) bind to the specific ligand with high affinity and mediate their transport into the cytoplasm via the cognate inner membrane ATP binding cassette (ABC) proteins. Because of very low sequence identities, understanding the structural basis of substrate recognition by SBPs has remained very challenging. A periplasmic glucose binding protein from Pseudomonas putida CSV86 (ppGBP) is found to be highly specific towards glucose with an affinity of ~0.3 µM and has very low specificity towards galactose. We have determined the first high resolution crystal structures of a periplasmic glucose binding protein (ppGBP from a Pseudomonas sp. The structures of ppGBP have been solved and refined as unliganded form (2.5 Å), as well as its complexes with glucose (1.25 Å) and galactose (1.8 Å). Asymmetric domain closure of ppGBP has been observed upon sugar binding. The structural analysis revealed that the sugars are bound to the protein mainly by hydrogen bonds and the loss of two strong hydrogen bonds between ppGBP and galactose compared to glucose is responsible for lowering its affinity towards galactose. Interestingly, ppGBP binds to only monosaccharide but it has the structural fold of an oligosaccharide binding protein. Our structural analysis indicates that during evolution, the sugar binding pocket of ppGBP may have undergone modulation to accommodate monosaccharides only.
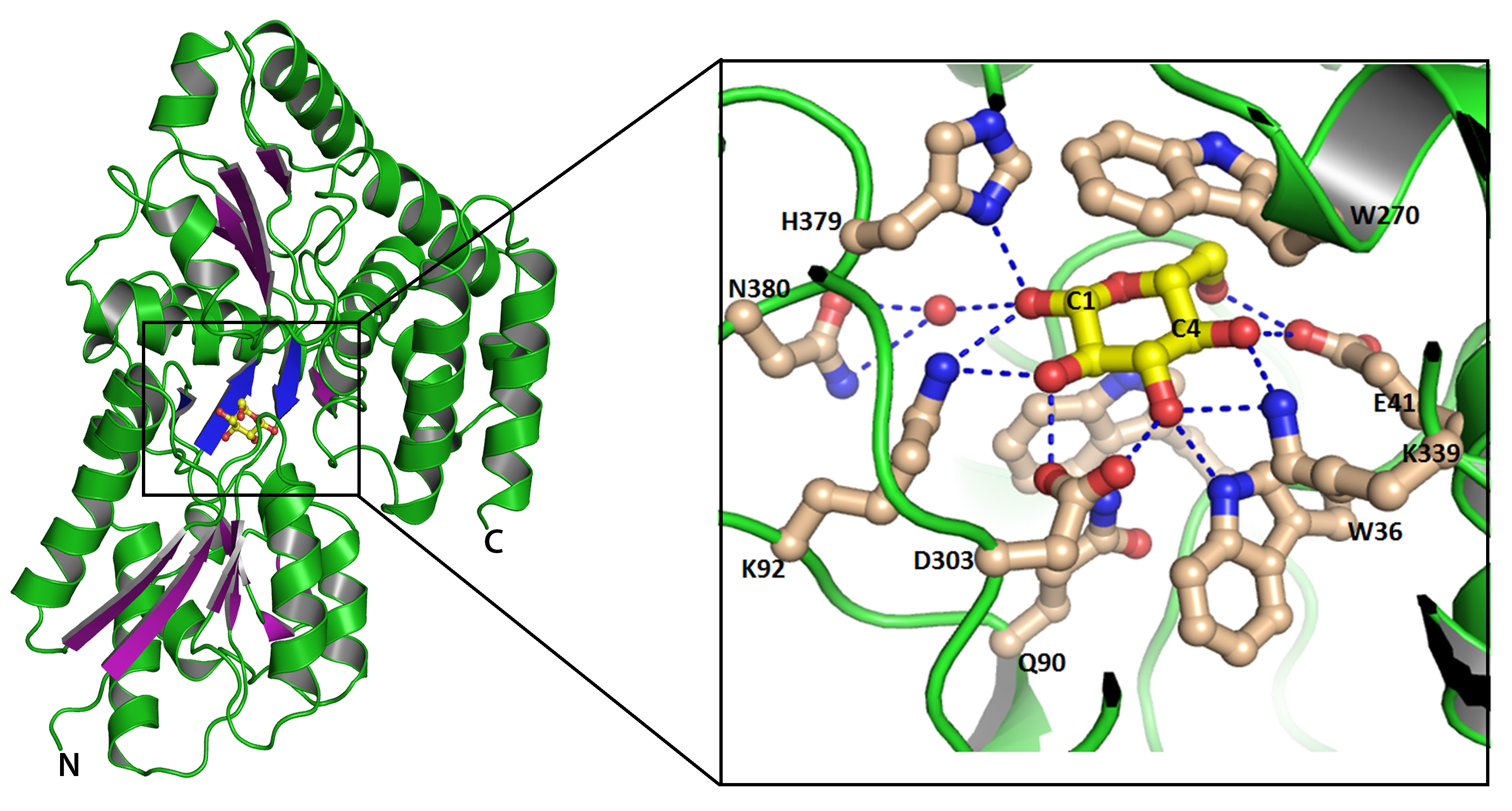 |
Overall structural fold of ppGBP represented as cartoon. The ß-sheets are shown in purple color to highlight their arrangements. Glucose is shown as ball and stick. Inset shows interactions of glucose with ppGBP. Carbon atoms of glucose and amino acid residues are shown as yellow and light-brown color, respectively. Hydrogen bonds are presented as dotted lines. |